

← Retour vers le trombinoscope
 Maître de conférences
Maître de conférencesMail :
Tel : 03 22 82 76 73
Structure-Activity Relationship of Benzophenazine Derivatives for Homogeneous and Heterogenized Photooxygenation Catalysis |
Comparative analysis of sulfated and sulfonated disaccharide analogs as TLR4 modulators and heparanase inhibitors |
Front Cover: The Mechanism of Lithium Zincate-Mediated I/Zn Exchange Revisited: A Computational Microsolvation Approach in THF (Eur. J. Org. Chem. 43/2023) |
The Mechanism of Lithium Zincate-Mediated I/Zn Exchange Revisited: A Computational Microsolvation Approach in THF** |
IGMPlot: A program to identify, characterize, and quantify molecular interactions |
New insight into atomic-level interpretation of interactions in molecules and reacting systems |
Photochemically Induced Hydrogen Atom Transfer and Intramolecular Radical Cyclization Reactions with Oxazolones |
Photocatalyzed Synthesis of 3-Substituted Phthalides: A Key Access to (±)-Herbaric Acid |
Impact of silica nanoparticles architectures on the photosensitization of O2 by immobilized Rose Bengal |
Photo-Catalyzed α-Arylation of Enol Acetate Using Recyclable Silica-Supported Heteroleptic and Homoleptic Copper(I) Photosensitizers |
Front Cover: Photocatalytic Radical Addition to Levoglucosenone (Eur. J. Org. Chem. 1/2022) |
Photocatalytic Radical Addition to Levoglucosenone |
Synthesis and characterization of polymethine dyes carrying thiobarbituric and carboxylic acid moieties |
Chapter Eight - Photochemical rearrangements in organic synthesis and the concept of the photon as a traceless reagent |
Studies on The Application of The Paternò-Büchi Reaction to The Synthesis of Novel Fluorinated Scaffolds |
Atomic Decomposition Scheme of Noncovalent Interactions Applied to Host–Guest Assemblies |
Photochemical Rearrangements in Heterocyclic Chemistry |
Front cover: Photochemical reactivity of phenyl (methyl-tetrazolyl) ketone – hydrogen atom transfer vs. electron transfer |
Photochemical reactivity of phenyl (methyl-tetrazolyl) ketone – hydrogen atom transfer vs. electron transfer |
Photochemically Induced Intramolecular Radical Cyclization Reactions with Imines |
The Independent Gradient Model: A New Approach for Probing Strong and Weak Interactions in Molecules from Wave Function Calculations |
Accurately extracting the signature of intermolecular interactions present in the NCI plot of the reduced density gradient versus electron density |
 Abstract Singlet oxygen is a powerful oxidant used in various applications, such as organic synthesis, medicine, and environmental remediation. Organic and inorganic photosensitizers are commonly used to generate this reactive species through energy transfer with the triplet ground state of oxygen. We describe here a series of novel benzophenazine derivatives as a promising class of photosensitizers for singlet oxygen photosensitization. In this study, we investigated the structure-activity relationship of these benzophenazine derivatives. Akin to a molecular compass, the southern fragment was first functionalized with either aromatic tertiary amines, alkyl tertiary amines, aromatic sulfur groups, alkyl sulfur groups, or cyclic ethers. Enhanced photophysical properties (in terms of triplet excited-state lifetime, absorption wavelength, triplet state energy, and O2 quenching capabilities) were obtained with cyclic ether and sulfur groups. Conversely, the presence of an amine moiety was detrimental to the photocatalysts. The western and northern fragments were also investigated and slightly undesirable to negligible changes in photophysical properties were observed. The most promising candidate was then immobilized on silica nanoparticles and its photoactivity was evaluated in the citronellol photooxidation reaction. A high NMR yield of 97 % in desired product was obtained, with only a slight decrease over several recycling runs (85 % in the fourth run). These results provide insights into the design of efficient photosensitizers for singlet oxygen generation and the development of heterogeneous systems.
Abstract Singlet oxygen is a powerful oxidant used in various applications, such as organic synthesis, medicine, and environmental remediation. Organic and inorganic photosensitizers are commonly used to generate this reactive species through energy transfer with the triplet ground state of oxygen. We describe here a series of novel benzophenazine derivatives as a promising class of photosensitizers for singlet oxygen photosensitization. In this study, we investigated the structure-activity relationship of these benzophenazine derivatives. Akin to a molecular compass, the southern fragment was first functionalized with either aromatic tertiary amines, alkyl tertiary amines, aromatic sulfur groups, alkyl sulfur groups, or cyclic ethers. Enhanced photophysical properties (in terms of triplet excited-state lifetime, absorption wavelength, triplet state energy, and O2 quenching capabilities) were obtained with cyclic ether and sulfur groups. Conversely, the presence of an amine moiety was detrimental to the photocatalysts. The western and northern fragments were also investigated and slightly undesirable to negligible changes in photophysical properties were observed. The most promising candidate was then immobilized on silica nanoparticles and its photoactivity was evaluated in the citronellol photooxidation reaction. A high NMR yield of 97 % in desired product was obtained, with only a slight decrease over several recycling runs (85 % in the fourth run). These results provide insights into the design of efficient photosensitizers for singlet oxygen generation and the development of heterogeneous systems. This article delves into the synthesis and biological evaluation of sulfated and sulfonated disaccharide analogs, exploring their interactions with toll-like receptor 4 (TLR4) and their potential as drug candidates for inflammatory diseases. A significant aspect of the study is the examination of these analogs as inhibitors of heparanase, an enzyme that cleaves glycosidic linkages in heparan sulfate, producing proinflammatory fragments that activate TLR4. The research presents a comparative analysis of sulfate and sulfonate groups in these compounds, evaluating their synthesis, biological activity, and specific roles in TLR4-mediated immune responses, with a particular focus on their ability to modulate heparanase activity. Compound 9 (6,6′-disulfonated disaccharide from methyl cellobioside) emerged as a potent TLR4 activator and a promising candidate for inflammatory drug development, exhibiting notable specificity and efficacy. The IC50 values for heparanase inhibition varied, highlighting distinct efficacy profiles for sulfonated and sulfated analogs, with cellobiose derivatives showing notable differences in inhibition capabilities.
This article delves into the synthesis and biological evaluation of sulfated and sulfonated disaccharide analogs, exploring their interactions with toll-like receptor 4 (TLR4) and their potential as drug candidates for inflammatory diseases. A significant aspect of the study is the examination of these analogs as inhibitors of heparanase, an enzyme that cleaves glycosidic linkages in heparan sulfate, producing proinflammatory fragments that activate TLR4. The research presents a comparative analysis of sulfate and sulfonate groups in these compounds, evaluating their synthesis, biological activity, and specific roles in TLR4-mediated immune responses, with a particular focus on their ability to modulate heparanase activity. Compound 9 (6,6′-disulfonated disaccharide from methyl cellobioside) emerged as a potent TLR4 activator and a promising candidate for inflammatory drug development, exhibiting notable specificity and efficacy. The IC50 values for heparanase inhibition varied, highlighting distinct efficacy profiles for sulfonated and sulfated analogs, with cellobiose derivatives showing notable differences in inhibition capabilities. The Front Cover shows the most favorable calculated transition state (TS) through which the I/Zn exchange reaction of 4-iodobenzyl mesylate with Et3ZnLi proceeds in THF. This TS was located through micro-solvation approach by contacting a doubly-THF solvated open zincate complex, identified as one of the 3 limit forms of the reagent, and the iodoaryl derivative. From this TS, the I/Zn exchange product is obtained via a 2 step-mechanism. First, the ArI substrate is converted into ArLi intermediate which then reacts with the remaining Et2Zn entity. More information can be found in the Research Article by Clément Denhez, Alexandre Vasseur, and co-workers.
The Front Cover shows the most favorable calculated transition state (TS) through which the I/Zn exchange reaction of 4-iodobenzyl mesylate with Et3ZnLi proceeds in THF. This TS was located through micro-solvation approach by contacting a doubly-THF solvated open zincate complex, identified as one of the 3 limit forms of the reagent, and the iodoaryl derivative. From this TS, the I/Zn exchange product is obtained via a 2 step-mechanism. First, the ArI substrate is converted into ArLi intermediate which then reacts with the remaining Et2Zn entity. More information can be found in the Research Article by Clément Denhez, Alexandre Vasseur, and co-workers. Abstract Lithium trialkylzincate-mediated I/Zn exchange reaction has been revisited computationally through a microsolvation approach. A never yet investigated iodoaryl derivative bearing a potential bulky para-directing group, namely 4-iodobenzyl mesylate, was considered as a substrate. THF as typical solvent and Et3ZnLi have also been considered for the first time in such a reaction. Three mechanistic pathways have been calculated, including (1) a literature-inspired pathway with full preservation of the synergic character of the reagent as well as a complementary mesylate group-directed pathway, (2) a THF-solvated open complex-promoted pathway and (3) an anionic pathway. While the anionic pathway appeared to be unlikely, pathway involving a THF-solvated open zincate complex turned out to be the most energetically favored. Equivalent thermodynamic profiles were found for both complementary pathways with preservation of the synergic character of the reagent, albeit a slight preference could be attributed to that occurring with initial chelation of Li to the mesylate group (OMs) through microsolvation approach. The I/Zn exchange was shown to proceed through a lithium-assisted aryl shuttle-like process. The iodoaryl substrate is first converted into ArLi intermediate which in turns reacts with the remaining diorganozinc reagent.
Abstract Lithium trialkylzincate-mediated I/Zn exchange reaction has been revisited computationally through a microsolvation approach. A never yet investigated iodoaryl derivative bearing a potential bulky para-directing group, namely 4-iodobenzyl mesylate, was considered as a substrate. THF as typical solvent and Et3ZnLi have also been considered for the first time in such a reaction. Three mechanistic pathways have been calculated, including (1) a literature-inspired pathway with full preservation of the synergic character of the reagent as well as a complementary mesylate group-directed pathway, (2) a THF-solvated open complex-promoted pathway and (3) an anionic pathway. While the anionic pathway appeared to be unlikely, pathway involving a THF-solvated open zincate complex turned out to be the most energetically favored. Equivalent thermodynamic profiles were found for both complementary pathways with preservation of the synergic character of the reagent, albeit a slight preference could be attributed to that occurring with initial chelation of Li to the mesylate group (OMs) through microsolvation approach. The I/Zn exchange was shown to proceed through a lithium-assisted aryl shuttle-like process. The iodoaryl substrate is first converted into ArLi intermediate which in turns reacts with the remaining diorganozinc reagent.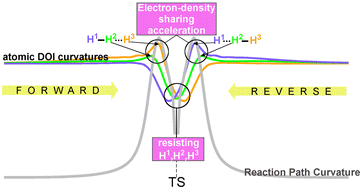 We hereby introduce the atomic degree of interaction (DOI), a new concept rooted in the electron density-based independent gradient model (IGM). Capturing any manifestation of electron density sharing around an atom, including covalent and non-covalent situations, this index reflects the attachment strength of an atom to its molecular neighbourhood. It is shown to be very sensitive to the local chemical environment of the atom. No significant correlation could be found between the atomic DOI and various other atomic properties, making this index a specific source of information. However, examining the simple H2 + H reacting system, a strong connection has been established between this electron density-based index and the scalar reaction path curvature, the cornerstone of the benchmark unified reaction valley approach (URVA). We observe that reaction path curvature peaks appear when atoms experience an acceleration phase of electron density sharing during the reaction, detected by peaks of the DOI second derivative either in the forward or reverse direction. This new IGM-DOI tool is only in its early stages, but it opens the way to an atomic-level interpretation of reaction phases. More generally, the IGM-DOI tool may also serve as an atomic probe of electronic structure changes of a molecule under the influence of physicochemical perturbations.
We hereby introduce the atomic degree of interaction (DOI), a new concept rooted in the electron density-based independent gradient model (IGM). Capturing any manifestation of electron density sharing around an atom, including covalent and non-covalent situations, this index reflects the attachment strength of an atom to its molecular neighbourhood. It is shown to be very sensitive to the local chemical environment of the atom. No significant correlation could be found between the atomic DOI and various other atomic properties, making this index a specific source of information. However, examining the simple H2 + H reacting system, a strong connection has been established between this electron density-based index and the scalar reaction path curvature, the cornerstone of the benchmark unified reaction valley approach (URVA). We observe that reaction path curvature peaks appear when atoms experience an acceleration phase of electron density sharing during the reaction, detected by peaks of the DOI second derivative either in the forward or reverse direction. This new IGM-DOI tool is only in its early stages, but it opens the way to an atomic-level interpretation of reaction phases. More generally, the IGM-DOI tool may also serve as an atomic probe of electronic structure changes of a molecule under the influence of physicochemical perturbations. Photochemically induced intramolecular hydrogen atom transfer in oxazolones is reported. An acetal or thioacetal function at the side chain acts as a hydrogen donor while the photochemical exited oxazolone is the acceptor. A one-step process─the electron and the proton are simultaneously transferred─is productive, while electron transfer followed by proton transfer is inefficient. Radical combination then takes place, leading to the formation of a C–C or C–N bond. The regioselectivity of the reaction is explained by the diradical/zwitterion dichotomy of radical intermediates at the singlet state. In the present case, the zwitterion structure plays a central role, and intramolecular electron transfer favors spin–orbit coupling and thus the intersystem crossing to the singlet state. The reaction of corresponding thioacetal derivatives is less efficient. In this case, photochemical electron transfer is competitive. The photoproducts resulting from C–C bond formation easily undergo stepwise thermal decarboxylation in which zwitterionic and polar transition states are involved. A computational study of this step has also been performed.
Photochemically induced intramolecular hydrogen atom transfer in oxazolones is reported. An acetal or thioacetal function at the side chain acts as a hydrogen donor while the photochemical exited oxazolone is the acceptor. A one-step process─the electron and the proton are simultaneously transferred─is productive, while electron transfer followed by proton transfer is inefficient. Radical combination then takes place, leading to the formation of a C–C or C–N bond. The regioselectivity of the reaction is explained by the diradical/zwitterion dichotomy of radical intermediates at the singlet state. In the present case, the zwitterion structure plays a central role, and intramolecular electron transfer favors spin–orbit coupling and thus the intersystem crossing to the singlet state. The reaction of corresponding thioacetal derivatives is less efficient. In this case, photochemical electron transfer is competitive. The photoproducts resulting from C–C bond formation easily undergo stepwise thermal decarboxylation in which zwitterionic and polar transition states are involved. A computational study of this step has also been performed. Abstract An efficient organophotocatalyzed protocol was developed for the preparation of 3-substituted phthalides. The presented transformation was performed under particularly mild conditions within 6 h and was ultimately applied to a precursor of the herbaric acid.
Abstract An efficient organophotocatalyzed protocol was developed for the preparation of 3-substituted phthalides. The presented transformation was performed under particularly mild conditions within 6 h and was ultimately applied to a precursor of the herbaric acid. Transforming light into chemical energy has been widely studied to design ecofriendly, cost-effective and sustainable synthetic approaches. The heterogenization of organic photocatalysts is a well-known strategy providing efficient solid supported catalysts. One of the most suitable solid as support is silica nanoparticles (NPs). This research note highlights the impact of silica nanoparticles morphology on a representative photochemical process that is the photooxidation of citronellol, involving oxygen sensitization. Three architectures of silica NPs were investigated: Nanospheres (NS), Core-Shell (CS) and Urchins (U). Their surfaces were functionalized with ammonium moieties allowing the immobilization of Rose Bengal (RB) by electrostatic interactions. The heterogenized RB on the various supports displayed different activities in the production of singlet oxygen, CS being the best support in terms of activity, recyclability and dye immobilization rate.
Transforming light into chemical energy has been widely studied to design ecofriendly, cost-effective and sustainable synthetic approaches. The heterogenization of organic photocatalysts is a well-known strategy providing efficient solid supported catalysts. One of the most suitable solid as support is silica nanoparticles (NPs). This research note highlights the impact of silica nanoparticles morphology on a representative photochemical process that is the photooxidation of citronellol, involving oxygen sensitization. Three architectures of silica NPs were investigated: Nanospheres (NS), Core-Shell (CS) and Urchins (U). Their surfaces were functionalized with ammonium moieties allowing the immobilization of Rose Bengal (RB) by electrostatic interactions. The heterogenized RB on the various supports displayed different activities in the production of singlet oxygen, CS being the best support in terms of activity, recyclability and dye immobilization rate. Abstract Earth-abundant photosensitizers are highly sought after for light-mediated applications, such as photoredox catalysis, depollution and energy conversion schemes. Homoleptic and heteroleptic copper(I) complexes are promising candidates in this field, as copper is abundant and the corresponding complexes are easily obtained in smooth conditions. However, some heteroleptic copper(I) complexes suffer from low (photo)stability that leads to the gradual formation of the corresponding homoleptic complex. Such degradation pathways are detrimental, especially when recyclability is desired. This study reports a novel approach for the heterogenization of homoleptic and heteroleptic Cu complexes on silica nanoparticles. In both cases, the photophysical properties upon surface immobilization were only slightly affected. Excited-state quenching with aryl diazonium derivatives occurred efficiently (108–1010 M−1 s−1) with heterogeneous and homogeneous photosensitizers. Moderate but almost identical yields were obtained for the α-arylation of enol acetate using the homoleptic complex in homogeneous or heterogeneous conditions. Importantly, the silica-supported photocatalysts were recycled with moderate loss in photoactivity over multiple experiments. Transient absorption spectroscopy confirmed that excited-state electron transfer occurred from the homogeneous and heterogeneous homoleptic copper(I) complexes to aryl diazonium derivatives, generating the corresponding copper(II) center that persisted for several hundreds of microseconds, compatible with photoredox catalysis applications.
Abstract Earth-abundant photosensitizers are highly sought after for light-mediated applications, such as photoredox catalysis, depollution and energy conversion schemes. Homoleptic and heteroleptic copper(I) complexes are promising candidates in this field, as copper is abundant and the corresponding complexes are easily obtained in smooth conditions. However, some heteroleptic copper(I) complexes suffer from low (photo)stability that leads to the gradual formation of the corresponding homoleptic complex. Such degradation pathways are detrimental, especially when recyclability is desired. This study reports a novel approach for the heterogenization of homoleptic and heteroleptic Cu complexes on silica nanoparticles. In both cases, the photophysical properties upon surface immobilization were only slightly affected. Excited-state quenching with aryl diazonium derivatives occurred efficiently (108–1010 M−1 s−1) with heterogeneous and homogeneous photosensitizers. Moderate but almost identical yields were obtained for the α-arylation of enol acetate using the homoleptic complex in homogeneous or heterogeneous conditions. Importantly, the silica-supported photocatalysts were recycled with moderate loss in photoactivity over multiple experiments. Transient absorption spectroscopy confirmed that excited-state electron transfer occurred from the homogeneous and heterogeneous homoleptic copper(I) complexes to aryl diazonium derivatives, generating the corresponding copper(II) center that persisted for several hundreds of microseconds, compatible with photoredox catalysis applications. The Front Cover shows the stereo and regio selective addition of radical species such as alkyl or formamidyl radicals to levoglucosenone. Levoglucosenone is a biobased platform chemical obtained from celluloses. The radical intermediates are generated by hydrogen atom transfer involving photocatalysis with tetra-n-butylammonium decatungstate (TBADT). Cover picture created with the help of Tony Leclet. More information can be found in the Article by N. Hoffmann et al.
The Front Cover shows the stereo and regio selective addition of radical species such as alkyl or formamidyl radicals to levoglucosenone. Levoglucosenone is a biobased platform chemical obtained from celluloses. The radical intermediates are generated by hydrogen atom transfer involving photocatalysis with tetra-n-butylammonium decatungstate (TBADT). Cover picture created with the help of Tony Leclet. More information can be found in the Article by N. Hoffmann et al. Abstract Using photocatalysis with tetra-n-butylammonium decatungstate (TBADT), alkanes, cyclic acetals, cyclic ethers, formamide and aldehydes were added in a stereoselective way to levoglucosenone (LGO). A hydrogen atom is transferred from the donor compound to the photochemically excited TBADT, and the resulting radicals add onto LGO in a stereoselective way. In the case of the addition of adamantane, two regio-isomers were obtained which form a crystalline solid solution. Cyrene™, obtained by hydrogenation of LGO, was added under the same conditions. In this case, only two of 32 possible isomers of the resulting Cyrene™ dimer were formed. The regio-selectivity of the HAT step is discussed in detail. For this purpose, bond dissociation energies and partial charges have been calculated. Transition state calculations of the radical addition to LGO explain the stereospecificity of this reaction step.
Abstract Using photocatalysis with tetra-n-butylammonium decatungstate (TBADT), alkanes, cyclic acetals, cyclic ethers, formamide and aldehydes were added in a stereoselective way to levoglucosenone (LGO). A hydrogen atom is transferred from the donor compound to the photochemically excited TBADT, and the resulting radicals add onto LGO in a stereoselective way. In the case of the addition of adamantane, two regio-isomers were obtained which form a crystalline solid solution. Cyrene™, obtained by hydrogenation of LGO, was added under the same conditions. In this case, only two of 32 possible isomers of the resulting Cyrene™ dimer were formed. The regio-selectivity of the HAT step is discussed in detail. For this purpose, bond dissociation energies and partial charges have been calculated. Transition state calculations of the radical addition to LGO explain the stereospecificity of this reaction step. An efficient synthesis of polymethine dyes carrying thiobarbituric and a carboxylic acid moiety has been developed. Such compounds play a key role in many photometric detections and quantifications of enzyme activities. In such tests, the metabolite of the enzyme activities is transformed into a β-dicarbonyl derivative. In the present study, this compound was prepared from furfural through organic synthesis. Its in situ transformation with thiobarbituric acid derivatives yields the target compounds on a gram-scale (0.4 to 0.6 g). A combined experimental and theoretical study of the photophysical properties of the synthesized compounds was carried out. Absorption and emission spectroscopy measurements highlighted a slight solvatochromism effect. The luminescence was quenched by molecular oxygen, indicating the partial triplet multiplicity character of the lowest excited state. Density Functional Theory (DFT) calculations have been applied for the evaluation of favoured conformations for these new compounds and the study of their optical properties. Within the Franck Condon principle, the vibrationally resolved electronic one-photon absorption spectrum has been simulated. This simulation shows the presence of a major band followed by a vibronic sideband, typical of organic chromophores in solution. The performed computational study revealed that the transition from the ground to the first excited electronic state has a π–π* character. Finally, TD–DFT energy level diagram calculations highlighted the presence of triplet states very close to the first singlet excited one, suggesting probable access to the triplet-excited state.
An efficient synthesis of polymethine dyes carrying thiobarbituric and a carboxylic acid moiety has been developed. Such compounds play a key role in many photometric detections and quantifications of enzyme activities. In such tests, the metabolite of the enzyme activities is transformed into a β-dicarbonyl derivative. In the present study, this compound was prepared from furfural through organic synthesis. Its in situ transformation with thiobarbituric acid derivatives yields the target compounds on a gram-scale (0.4 to 0.6 g). A combined experimental and theoretical study of the photophysical properties of the synthesized compounds was carried out. Absorption and emission spectroscopy measurements highlighted a slight solvatochromism effect. The luminescence was quenched by molecular oxygen, indicating the partial triplet multiplicity character of the lowest excited state. Density Functional Theory (DFT) calculations have been applied for the evaluation of favoured conformations for these new compounds and the study of their optical properties. Within the Franck Condon principle, the vibrationally resolved electronic one-photon absorption spectrum has been simulated. This simulation shows the presence of a major band followed by a vibronic sideband, typical of organic chromophores in solution. The performed computational study revealed that the transition from the ground to the first excited electronic state has a π–π* character. Finally, TD–DFT energy level diagram calculations highlighted the presence of triplet states very close to the first singlet excited one, suggesting probable access to the triplet-excited state. Abstract In the context of new scaffolds obtained by photochemical reactions, Paternò-Büchi reactions between heteroaromatic, trifluoromethylphenyl ketone and electron rich alkenes to give oxetanes are described. A comprehensive study has then been carried out on the reaction of aromatic ketones with fluorinated alkenes. Depending on the substitution pattern at the oxetane ring, a metathesis reaction is described as a minor side process to give mono fluorinated alkenes. Overall, this last reaction corresponds to a photo-Wittig reaction and yield amid isosteres. In order to explain the uncommon regioselectivity of the Paternò-Büchi reaction with these alkenes, electrostatic-potential derived charges (ESP) have been determined. In a second computational study, the relative stabilities of the typical 1,4-diradical intermediates of the Paternò-Büchi reaction have been determined. The results well explain the regioselectivity. Further transformations of the oxetanes or previous functionalization of the fluoroalkenes open perspectives for oxetanes as core structures for biologically active compounds.
Abstract In the context of new scaffolds obtained by photochemical reactions, Paternò-Büchi reactions between heteroaromatic, trifluoromethylphenyl ketone and electron rich alkenes to give oxetanes are described. A comprehensive study has then been carried out on the reaction of aromatic ketones with fluorinated alkenes. Depending on the substitution pattern at the oxetane ring, a metathesis reaction is described as a minor side process to give mono fluorinated alkenes. Overall, this last reaction corresponds to a photo-Wittig reaction and yield amid isosteres. In order to explain the uncommon regioselectivity of the Paternò-Büchi reaction with these alkenes, electrostatic-potential derived charges (ESP) have been determined. In a second computational study, the relative stabilities of the typical 1,4-diradical intermediates of the Paternò-Büchi reaction have been determined. The results well explain the regioselectivity. Further transformations of the oxetanes or previous functionalization of the fluoroalkenes open perspectives for oxetanes as core structures for biologically active compounds. The design of novel stimuli-responsive supramolecular systems based on host–guest chemistry implies a thorough understanding of the noncovalent interactions involved. In this regard, some computational tools enabling the extraction of the noncovalent signatures from local descriptors based on the electron density have been previously proposed. Although very useful to detect the existence of such interactions, these analyses provide only a semi-quantitative description, which represents a limitation. In this work, we present a novel computational tool based on the local atomic descriptor IGM-δginter/At, which is able to decompose the fragment interaction into atomic contributions. Then, the role played by each atom in the formation of the host–guest assembly is quantified by an integrated Δginter/At score. Herein, we apply the IGM-Δginter/At approach to some challenging systems, including multimetallic arrays, buckycatchers, and organic assemblies. These systems exhibit unique structural features that make it difficult to determine the host/guest atoms that contribute the most to the guest encapsulation. Here, the Δginter/At score proves to be an appealing tool to shed light on the guest accommodation on a per-atom basis and could be useful in the rational design of more selective target agents. We strongly believe that this novel approach will be useful for experimental teams devoted to the synthesis of supramolecular systems based on host–guest chemistry.
The design of novel stimuli-responsive supramolecular systems based on host–guest chemistry implies a thorough understanding of the noncovalent interactions involved. In this regard, some computational tools enabling the extraction of the noncovalent signatures from local descriptors based on the electron density have been previously proposed. Although very useful to detect the existence of such interactions, these analyses provide only a semi-quantitative description, which represents a limitation. In this work, we present a novel computational tool based on the local atomic descriptor IGM-δginter/At, which is able to decompose the fragment interaction into atomic contributions. Then, the role played by each atom in the formation of the host–guest assembly is quantified by an integrated Δginter/At score. Herein, we apply the IGM-Δginter/At approach to some challenging systems, including multimetallic arrays, buckycatchers, and organic assemblies. These systems exhibit unique structural features that make it difficult to determine the host/guest atoms that contribute the most to the guest encapsulation. Here, the Δginter/At score proves to be an appealing tool to shed light on the guest accommodation on a per-atom basis and could be useful in the rational design of more selective target agents. We strongly believe that this novel approach will be useful for experimental teams devoted to the synthesis of supramolecular systems based on host–guest chemistry. Heterocyclic compounds play an important role in many domains of chemistry. They are important structural elements in bioactive compounds. Photochemical reactions enable transformations of such compounds in a very convenient way. In many cases no chemical reagent is used. Members of one compound family can be transformed into members of another one. Three important types of photochemical rearrangements with heterocyclic compounds are discussed: Photochemical heteroatom isomerization involving heteroatoms and substituents, photochemical reactions involving hydrogen atom transfer (HAT) and photochemical electrocyclization.
Heterocyclic compounds play an important role in many domains of chemistry. They are important structural elements in bioactive compounds. Photochemical reactions enable transformations of such compounds in a very convenient way. In many cases no chemical reagent is used. Members of one compound family can be transformed into members of another one. Three important types of photochemical rearrangements with heterocyclic compounds are discussed: Photochemical heteroatom isomerization involving heteroatoms and substituents, photochemical reactions involving hydrogen atom transfer (HAT) and photochemical electrocyclization.
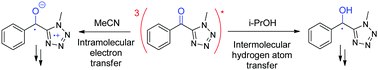 Phenyl (methyl-tetrazolyl) ketone (1) is a synthesis intermediate of tetrazolyloxime fungicides and can also be generated upon their irradiation. Its photolysis is highly solvent-dependent, which prompted us to investigate the reaction mechanism more deeply. The nanosecond laser flash photolysis of 1 yielded the triplet excited state (λmax = 390/570 nm) immediately after the pulse. This was later converted into different secondary species that were identified using their specific reactivity as well as product studies. The ketyl radical (λmax = 315/475 nm) was generated in less than 0.02 μs in a good H-donor solvent such as 2-propanol and in around 0.06 μs in cyclohexane, a medium H-donor solvent. In 2-propanol, ketyl radicals decayed by a second order reaction to yield pinacol (yield 45%); in contrast, in cyclohexane, they decayed by a second order reaction in the bulk, leading to the formation of pinacol (yield 21%), and by recombination with the cyclohexyl radical in the cage in an apparent first order reaction to generate an adduct (yield 10%). In a polar and non H-atom donor solvent such as acetonitrile, the zwitterionic diradical (λmax = 460 nm) was formed in 0.6 μs with the final formation of an atypical dimer. Thus, two mechanisms of hydrogen atom transfer were observed. In the polar acetonitrile solvent, a two-step process occurred, where an electron was transferred first, followed by a proton. In the less polar 2-propanol and non-polar cyclohexane solvents, a one-step process occurred, where an electron and a proton were simultaneously transferred.
Phenyl (methyl-tetrazolyl) ketone (1) is a synthesis intermediate of tetrazolyloxime fungicides and can also be generated upon their irradiation. Its photolysis is highly solvent-dependent, which prompted us to investigate the reaction mechanism more deeply. The nanosecond laser flash photolysis of 1 yielded the triplet excited state (λmax = 390/570 nm) immediately after the pulse. This was later converted into different secondary species that were identified using their specific reactivity as well as product studies. The ketyl radical (λmax = 315/475 nm) was generated in less than 0.02 μs in a good H-donor solvent such as 2-propanol and in around 0.06 μs in cyclohexane, a medium H-donor solvent. In 2-propanol, ketyl radicals decayed by a second order reaction to yield pinacol (yield 45%); in contrast, in cyclohexane, they decayed by a second order reaction in the bulk, leading to the formation of pinacol (yield 21%), and by recombination with the cyclohexyl radical in the cage in an apparent first order reaction to generate an adduct (yield 10%). In a polar and non H-atom donor solvent such as acetonitrile, the zwitterionic diradical (λmax = 460 nm) was formed in 0.6 μs with the final formation of an atypical dimer. Thus, two mechanisms of hydrogen atom transfer were observed. In the polar acetonitrile solvent, a two-step process occurred, where an electron was transferred first, followed by a proton. In the less polar 2-propanol and non-polar cyclohexane solvents, a one-step process occurred, where an electron and a proton were simultaneously transferred. The photochemically induced intramolecular hydrogen abstraction or hydrogen atom transfer in cyclic imines 8a,b followed by a cyclization is investigated. Two types of products are observed, one resulting from the formation of a C–C bond, the other from the formation of a C–N bond. A computational study reveals that hydrogen is exclusively transferred to the imine nitrogen leading to a triplet diradical intermediate. After intersystem crossing, the resulting zwitterionic intermediate undergoes cyclization leading to the final product.
The photochemically induced intramolecular hydrogen abstraction or hydrogen atom transfer in cyclic imines 8a,b followed by a cyclization is investigated. Two types of products are observed, one resulting from the formation of a C–C bond, the other from the formation of a C–N bond. A computational study reveals that hydrogen is exclusively transferred to the imine nitrogen leading to a triplet diradical intermediate. After intersystem crossing, the resulting zwitterionic intermediate undergoes cyclization leading to the final product. Abstract Extraction of the chemical interaction signature from local descriptors based on electron density (ED) is still a fruitful field of development in chemical interpretation. In a previous work that used promolecular ED (frozen ED), the new descriptor, , was defined. It represents the difference between a virtual upper limit of the ED gradient ( , IGM=independent gradient model) that represents a noninteracting system and the true ED gradient ( ). It can be seen as a measure of electron sharing brought by ED contragradience. A compelling feature of this model is to provide an automatic workflow that extracts the signature of interactions between selected groups of atoms. As with the noncovalent interaction (NCI) approach, it provides chemists with a visual understanding of the interactions present in chemical systems. is achieved simply by using absolute values upon summing the individual gradient contributions that make up the total ED gradient. Hereby, we extend this model to relaxed ED calculated from a wave function. To this end, we formulated gradient-based partitioning (GBP) to assess the contribution of each orbital to the total ED gradient. We highlight these new possibilities across two prototypical examples of organic chemistry: the unconventional hexamethylbenzene dication, with a hexa-coordinated carbon atom, and β-thioaminoacrolein. It will be shown how a bond-by-bond picture can be obtained from a wave function, which opens the way to monitor specific interactions along reaction paths.
Abstract Extraction of the chemical interaction signature from local descriptors based on electron density (ED) is still a fruitful field of development in chemical interpretation. In a previous work that used promolecular ED (frozen ED), the new descriptor, , was defined. It represents the difference between a virtual upper limit of the ED gradient ( , IGM=independent gradient model) that represents a noninteracting system and the true ED gradient ( ). It can be seen as a measure of electron sharing brought by ED contragradience. A compelling feature of this model is to provide an automatic workflow that extracts the signature of interactions between selected groups of atoms. As with the noncovalent interaction (NCI) approach, it provides chemists with a visual understanding of the interactions present in chemical systems. is achieved simply by using absolute values upon summing the individual gradient contributions that make up the total ED gradient. Hereby, we extend this model to relaxed ED calculated from a wave function. To this end, we formulated gradient-based partitioning (GBP) to assess the contribution of each orbital to the total ED gradient. We highlight these new possibilities across two prototypical examples of organic chemistry: the unconventional hexamethylbenzene dication, with a hexa-coordinated carbon atom, and β-thioaminoacrolein. It will be shown how a bond-by-bond picture can be obtained from a wave function, which opens the way to monitor specific interactions along reaction paths. An electron density (ED)-based methodology is developed for the automatic identification of intermolecular interactions using pro-molecular density. The expression of the ED gradient in terms of atomic components furnishes the basis for the Independent Gradient Model (IGM). This model leads to a density reference for non interacting atoms/fragments where the atomic densities are added whilst their interaction turns off. Founded on this ED reference function that features an exponential decay also in interference regions, IGM model provides a way to identify and quantify the net ED gradient attenuation due to interactions. Using an intra/inter uncoupling scheme, a descriptor (δginter) is then derived that uniquely defines intermolecular interaction regions. An attractive feature of the IGM methodology is to provide a workflow that automatically generates data composed solely of intermolecular interactions for drawing the corresponding 3D isosurface representations.
An electron density (ED)-based methodology is developed for the automatic identification of intermolecular interactions using pro-molecular density. The expression of the ED gradient in terms of atomic components furnishes the basis for the Independent Gradient Model (IGM). This model leads to a density reference for non interacting atoms/fragments where the atomic densities are added whilst their interaction turns off. Founded on this ED reference function that features an exponential decay also in interference regions, IGM model provides a way to identify and quantify the net ED gradient attenuation due to interactions. Using an intra/inter uncoupling scheme, a descriptor (δginter) is then derived that uniquely defines intermolecular interaction regions. An attractive feature of the IGM methodology is to provide a workflow that automatically generates data composed solely of intermolecular interactions for drawing the corresponding 3D isosurface representations.